










Hesablama kimyası metodu elektron struktur hesablamalarından daha çox məlumatı sıxışdıra bilər
Köhnə günlərdə – həqiqətən köhnə günlərdə – materialların layihələndirilməsi işi çox çətin idi. Tədqiqatçılar 1000-dən çox il ərzində qurğuşun, civə və kükürd kimi şeyləri birləşdirərək qızıl düzəltməyə çalışdılar. Hətta Tycho Brahe, Robert Boyle və Isaac Newton kimi məşhur elm adamları kimyagərlik dediyimiz nəticəsiz cəhddə əllərini sınadılar.
Materialşünaslıq, əlbəttə ki, uzun bir yol keçmişdir. Son 150 il ərzində tədqiqatçılar müxtəlif elementlərin fərqli xüsusiyyətlərə malik olduğunu və birinin sehrli şəkildə digərinə çevrilə bilməyəcəyini söyləyən elementlərin dövri cədvəlindən faydalanıblar. Üstəlik, son onillikdə maşın öyrənmə vasitələri müxtəlif molekulların və maddələrin quruluşunu və fiziki xassələrini müəyyən etmək qabiliyyətimizi xeyli artırdı.
Tokio Elektrik Enerjisi Şirkətinin MIT-də Nüvə Mühəndisliyi professoru və materialşünaslıq və mühəndislik professoru Ju Linin rəhbərlik etdiyi bir qrup tərəfindən aparılan yeni tədqiqat materialların dizaynını asanlaşdıra biləcək imkanlarda böyük sıçrayış vəd edir. Onların araşdırmalarının nəticələri Nature Computational Science jurnalında dərc olunub .
Hal-hazırda, molekulyar sistemləri xarakterizə etmək üçün istifadə olunan maşın öyrənmə modellərinin əksəriyyəti elektron sıxlığının paylanmasına baxaraq molekulun və ya kristalın ümumi enerjisini təyin etmək üçün kvant mexaniki bir yanaşma təklif edən sıxlıq funksional nəzəriyyəsinə (DFT) əsaslanır. -bu, əsasən, molekulun yaxınlığında kosmosda hər bir verilmiş nöqtə ətrafında vahid həcmdə yerləşən elektronların orta sayıdır. (60 il əvvəl bu nəzəriyyəni icad edən Valter Kohn 1998-ci ildə kimya üzrə Nobel mükafatı aldı.)
Metod çox uğurlu olsa da, Linin fikrincə, onun bəzi çatışmazlıqları var: “Birincisi, dəqiqlik eyni dərəcədə böyük deyil. İkincisi, o, sizə yalnız bir şeyi deyir: molekulyar sistemin ən aşağı ümumi enerjisi.”
https://googleads.g.doubleclick.net/pagead/ads?client=ca-pub-0536483524803400&output=html&h=188&slotname=8188791252&adk=1687169288&adf=4054963813&pi=t.ma~as.8188791252&w=750&abgtt=6&fwrn=4&lmt=1736921258&rafmt=11&format=750×188&url=https%3A%2F%2Fphys.org%2Fnews%2F2025-01-chemistry-method-electronic.html&wgl=1&uach=WyJXaW5kb3dzIiwiMTkuMC4wIiwieDg2IiwiIiwiMTMxLjAuNjc3OC4yNjYiLG51bGwsMCxudWxsLCI2NCIsW1siR29vZ2xlIENocm9tZSIsIjEzMS4wLjY3NzguMjY2Il0sWyJDaHJvbWl1bSIsIjEzMS4wLjY3NzguMjY2Il0sWyJOb3RfQSBCcmFuZCIsIjI0LjAuMC4wIl1dLDBd&dt=1736921257937&bpp=1&bdt=63&idt=99&shv=r20250113&mjsv=m202501140101&ptt=9&saldr=aa&abxe=1&cookie=ID%3Df22668bce9793ae4%3AT%3D1735196613%3ART%3D1736921204%3AS%3DALNI_Mb4Xpwl1SO1AcvqroR6xccDm_sheQ&gpic=UID%3D00000f7c5320f40b%3AT%3D1735196613%3ART%3D1736921204%3AS%3DALNI_Mb1dz_DHiT2yDzXLMaB9CDkQl4XGg&eo_id_str=ID%3Dcdf7f2f01784f52d%3AT%3D1735196613%3ART%3D1736921204%3AS%3DAA-Afjb8kbeupLLyQ0QHQmZxpM4v&prev_fmts=0x0&nras=1&correlator=3321258959019&frm=20&pv=1&rplot=4&u_tz=240&u_his=3&u_h=1080&u_w=1920&u_ah=1032&u_aw=1920&u_cd=24&u_sd=1&dmc=8&adx=447&ady=2095&biw=1903&bih=945&scr_x=0&scr_y=0&eid=95349948%2C31088039%2C31089542%2C31089684%2C95344791%2C95350243%2C31089762%2C95347432%2C95340253%2C95340255&oid=2&pvsid=4211965042768516&tmod=1716569963&uas=0&nvt=1&ref=https%3A%2F%2Fphys.org%2F&fc=1920&brdim=0%2C0%2C0%2C0%2C1920%2C0%2C1920%2C1032%2C1920%2C945&vis=1&rsz=%7C%7CpeEbr%7C&abl=CS&pfx=0&fu=128&bc=31&bz=1&td=1&tdf=2&psd=W251bGwsbnVsbCxudWxsLDNd&nt=1&ifi=2&uci=a!2&btvi=1&fsb=1&dtd=102
Xilasetmə üçün ‘Cütlər terapiyası’
Onun komandası indi fərqli hesablama kimyası texnikasına güvənir, eyni zamanda birləşdirilmiş klaster nəzəriyyəsi və ya CCSD(T) kimi tanınan kvant mexanikasından əldə edilir.
“Bu, kvant kimyasının qızıl standartıdır” deyə Li şərh edir.
CCSD(T) hesablamalarının nəticələri DFT hesablamalarından əldə etdiyinizdən daha dəqiqdir və onlar hazırda təcrübələrdən əldə edilənlər qədər etibarlı ola bilər. Problem ondadır ki, bu hesablamaların kompüterdə aparılması çox ləng gedir, o deyir ki, “və miqyası pisdir: sistemdəki elektronların sayını iki dəfə artırsanız, hesablamalar 100 dəfə bahalaşar”.
Bu səbəbdən, CCSD(T) hesablamaları adətən az sayda atomlu molekullarla məhdudlaşır – təxminən 10. Bundan çox kənarda olan hər şey sadəcə çox uzun çəkər.
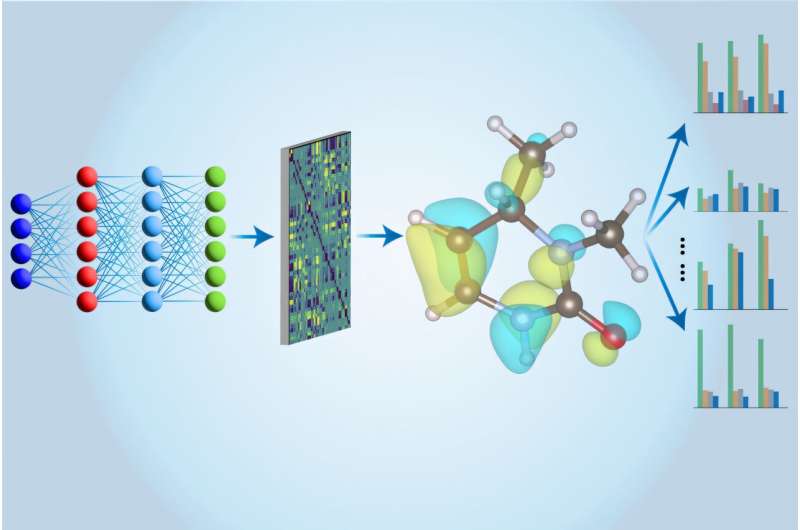
Məhz burada maşın öyrənməsi işə düşür. CCSD(T) hesablamaları ilk olaraq adi kompüterlərdə aparılır və nəticələr daha sonra Li və onun həmkarları tərəfindən xüsusi olaraq hazırlanmış yeni arxitekturaya malik neyron şəbəkəsini hazırlamaq üçün istifadə olunur. Təlimdən sonra neyron şəbəkə yaxınlaşma üsullarından istifadə edərək eyni hesablamaları daha sürətli yerinə yetirə bilər. Üstəlik, onların neyron şəbəkəsi modeli molekul haqqında onun enerjisindən daha çox məlumat əldə edə bilər.
MIT Ph.D Hao Tang deyir: “Əvvəlki işlərdə insanlar müxtəlif xüsusiyyətləri qiymətləndirmək üçün bir neçə fərqli modeldən istifadə etdilər”. materialşünaslıq və mühəndislik ixtisası üzrə tələbə. “Burada bütün bu xüsusiyyətləri qiymətləndirmək üçün yalnız bir modeldən istifadə edirik, buna görə də biz bunu “çox vəzifəli” yanaşma adlandırırıq.”
Gündəlik anlayışlar üçün Phys.org-a etibar edən 100.000-dən çox abunəçi ilə elm, texnologiya və kosmosda ən son yenilikləri kəşf edin . Pulsuz xəbər bülleteni üçün qeydiyyatdan keçin və mühüm nailiyyətlər, innovasiyalar və tədqiqatlar haqqında gündəlik və ya həftəlik yeniliklər əldə edin .Abunə ol
“Çox vəzifəli Elektron Hamilton şəbəkəsi” və ya MEHnet, dipol və dördqütblü momentlər, elektron qütbləşmə qabiliyyəti və optik həyəcan boşluğu kimi bir sıra elektron xassələri işıqlandırır. əsas vəziyyətindən ən aşağı həyəcanlı vəziyyətə.
Tang izah edir ki, “həyəcan boşluğu materialların optik xüsusiyyətlərinə təsir edir, çünki o, molekul tərəfindən udula bilən işığın tezliyini təyin edir.
Onların CCSD tərəfindən öyrədilmiş modelinin digər üstünlüyü ondan ibarətdir ki, o, təkcə yerüstü vəziyyətlərin deyil, həm də həyəcanlı vəziyyətlərin xüsusiyyətlərini aşkar edə bilər. Model həmçinin molekulun tərkibindəki atomların vibrasiyalarının bir-birinə bağlandığı və müxtəlif kollektiv davranışlara səbəb olduğu titrəmə xassələri ilə əlaqəli molekulun infraqırmızı udma spektrini proqnozlaşdıra bilər.
Onların yanaşmasının gücü şəbəkə arxitekturasına çox borcludur. MIT köməkçisi professor Tess Smidtin işinə əsaslanaraq, komanda E(3)-ekvivariant qrafik neyron şəbəkəsindən istifadə edir , Tang deyir ki, burada qovşaqlar atomları, qovşaqları birləşdirən kənarlar isə bir-birləri ilə əlaqəni təmsil edir. Biz həmçinin fizika prinsiplərini özündə birləşdirən fərdiləşdirilmiş alqoritmlərdən istifadə edirik. kvant mexanikası – birbaşa bizim modelimizə.”
https://googleads.g.doubleclick.net/pagead/ads?gdpr=0&us_privacy=1—&gpp_sid=-1&client=ca-pub-0536483524803400&output=html&h=188&slotname=8188791252&adk=1687169288&adf=809300024&pi=t.ma~as.8188791252&w=750&abgtt=6&fwrn=4&lmt=1736921446&rafmt=11&format=750×188&url=https%3A%2F%2Fphys.org%2Fnews%2F2025-01-chemistry-method-electronic.html&wgl=1&uach=WyJXaW5kb3dzIiwiMTkuMC4wIiwieDg2IiwiIiwiMTMxLjAuNjc3OC4yNjYiLG51bGwsMCxudWxsLCI2NCIsW1siR29vZ2xlIENocm9tZSIsIjEzMS4wLjY3NzguMjY2Il0sWyJDaHJvbWl1bSIsIjEzMS4wLjY3NzguMjY2Il0sWyJOb3RfQSBCcmFuZCIsIjI0LjAuMC4wIl1dLDBd&dt=1736921258061&bpp=1&bdt=187&idt=1&shv=r20250113&mjsv=m202501140101&ptt=9&saldr=aa&abxe=1&cookie=ID%3Df22668bce9793ae4%3AT%3D1735196613%3ART%3D1736921204%3AS%3DALNI_Mb4Xpwl1SO1AcvqroR6xccDm_sheQ&gpic=UID%3D00000f7c5320f40b%3AT%3D1735196613%3ART%3D1736921204%3AS%3DALNI_Mb1dz_DHiT2yDzXLMaB9CDkQl4XGg&eo_id_str=ID%3Dcdf7f2f01784f52d%3AT%3D1735196613%3ART%3D1736921204%3AS%3DAA-Afjb8kbeupLLyQ0QHQmZxpM4v&prev_fmts=0x0%2C750x188%2C1903x945%2C1005x124&nras=3&correlator=3321258959019&frm=20&pv=1&rplot=4&u_tz=240&u_his=4&u_h=1080&u_w=1920&u_ah=1032&u_aw=1920&u_cd=24&u_sd=1&dmc=8&adx=447&ady=3972&biw=1903&bih=945&scr_x=0&scr_y=195&eid=95349948%2C31088039%2C31089542%2C31089684%2C95344791%2C95350243%2C31089762%2C95347432%2C95340253%2C95340255&oid=2&psts=AOrYGsk9fB2OnbpeBDT0-K82d_RxARHSzrUKD4NYVGXvzZBtjkuoHkqxNtsGLCZcbnmi-q5WPzKwwDC4sHF5WDCJN_tGRWpy%2CAOrYGsk0HPALopRjaWzi2RNdopWuGiMcMWqLg6dF6QJ7um068PAKmnh3KNhHYx7DJe8L16Av4iLLBOF0rKVyefoptJjXsBTgVg0WWDTEUk37ErEyw6tNzQ&pvsid=4211965042768516&tmod=1716569963&uas=3&nvt=1&ref=https%3A%2F%2Fphys.org%2F&fc=1920&brdim=0%2C0%2C0%2C0%2C1920%2C0%2C1920%2C1032%2C1920%2C945&vis=1&rsz=%7C%7CpeEbr%7C&abl=CS&pfx=0&fu=128&bc=31&bz=1&td=1&tdf=2&psd=W251bGwsbnVsbCxudWxsLDNd&nt=1&ifi=5&uci=a!5&btvi=3&fsb=1&dtd=M
Test, 1, 2 3
Məlum karbohidrogen molekullarının təhlili üzərində sınaqdan keçirildikdə, model DFT analoqlarını üstələdi və nəşr olunmuş ədəbiyyatdan götürülmüş eksperimental nəticələrə yaxından uyğun gəldi.
Bu tədqiqatın bir hissəsi olmayan Şarlottadakı Şimali Karolina Universitetində materialların kəşfiyyatı üzrə mütəxəssis olan Qiang Zhu indiyə qədər əldə edilənlərdən təsirlənir.
“Onların metodu mövcud modellərlə müqayisədə daha yüksək dəqiqlik və hesablama səmərəliliyinə nail olmaqla, kiçik verilənlər bazası ilə effektiv təlimə imkan verir” dedi. “Bu, hesablama kimyası və dərin öyrənmə arasında güclü sinerji nümayiş etdirən , daha dəqiq və genişlənə bilən elektron struktur metodlarının işlənib hazırlanması üçün təzə ideyalar təklif edən maraqlı işdir .”
MIT-ə əsaslanan qrup öz modelini əvvəlcə kiçik, qeyri-metal elementlərə – hidrogen, karbon, azot, oksigen və flüora tətbiq etdi, onlardan üzvi birləşmələr hazırlana bilər – və o vaxtdan daha ağır elementləri araşdırmağa keçdi: silisium, fosfor, kükürd, xlor və hətta platin. Kiçik molekullar üzərində öyrədildikdən sonra model daha böyük və daha böyük molekullara ümumiləşdirilə bilər.
Li deyir: “Əvvəllər əksər hesablamalar DFT ilə yüzlərlə atomun və CCSD(T) hesablamaları ilə onlarla atomun təhlili ilə məhdudlaşırdı”. “İndi biz minlərlə atomun və nəhayət, bəlkə də on minlərlə atomun idarə edilməsindən danışırıq.”
Hələlik, tədqiqatçılar hələ də məlum molekulları qiymətləndirirlər, lakin model əvvəllər görülməmiş molekulları xarakterizə etmək, həmçinin müxtəlif molekullardan ibarət hipotetik materialların xüsusiyyətlərini proqnozlaşdırmaq üçün istifadə edilə bilər. “İdeya nəzəri alətlərimizdən istifadə edərək, müəyyən meyarlar toplusuna cavab verən perspektivli namizədləri yoxlamaq üçün eksperimentalistə təklif etməzdən əvvəl onları seçməkdir” dedi Tang.
Bütün bunlar proqramlara aiddir
İrəliyə baxaraq, Zhu mümkün tətbiqlər haqqında nikbindir. “Bu yanaşma yüksək məhsuldarlıqlı molekulyar skrininq üçün potensiala malikdir” deyir. “Bu, kimyəvi dəqiqliyə nail olmağın arzu olunan xüsusiyyətlərə malik yeni molekulları və materialları müəyyən etmək üçün vacib ola biləcəyi bir vəzifədir.”
Onlar bəlkə də on minlərlə atomu olan böyük molekulları təhlil etmək qabiliyyətini nümayiş etdirdikdən sonra Li deyir ki, dərman dizaynında və ya yarımkeçirici cihazlarda istifadə oluna biləcək “yeni polimerlər və ya materiallar icad edə bilməliyik”. Daha ağır keçid metal elementlərinin tədqiqi batareyalar üçün yeni materialların meydana çıxmasına səbəb ola bilər – hazırda kəskin ehtiyac sahəsi.
Gələcək, Li onu gördüyü kimi, açıqdır.
“Artıq bu, təkcə bir sahəyə aid deyil,” o deyir. “Bizim ambisiyamız, nəhayət, bütün dövri cədvəli CCSD(T) səviyyəli dəqiqliklə əhatə etməkdir, lakin DFT-dən daha aşağı hesablama xərcləri ilə. Bu, bizə kimya, biologiya və materialşünaslıqda geniş spektrli problemləri həll etməyə imkan verməlidir. Hazırda bu diapazonun nə qədər geniş ola biləcəyini bilmək çətindir”.
Daha çox məlumat: Hao Tang et al, Çox tapşırıqlı öyrənmə ilə molekulyar elektron strukturlar üçün birləşdirilmiş çoxluq dəqiqliyinə yaxınlaşma, Təbiət Hesablama Elmi (2024). DOI: 10.1038/s43588-024-00747-9
Jurnal məlumatı: Nature Computational Science
Massaçusets Texnologiya İnstitutu tərəfindən təmin edilmişdir

