










Maşın öyrənmə tətbiqi qabaqcıl kimyəvi proqnozları daha asan və sürətli edir, heç bir dərin proqramlaşdırma bacarığı tələb olunmur
Danielle Randall Doughty, Massaçusets Texnologiya İnstitutu
Stefani Baum tərəfindən redaktə edilmişdir , Andrew Zinin tərəfindən nəzərdən keçirilmişdir
Redaktorların qeydləriKredit: Kimyəvi İnformasiya və Modelləşdirmə jurnalı (2025). DOI: 10.1021/acs.jcim.5c00516
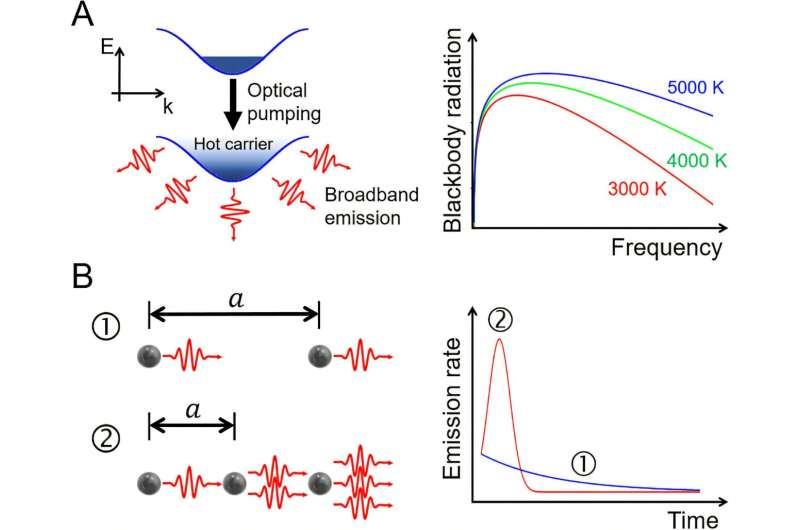
Əksər kimya tədqiqatçılarının ümumi, əsas məqsədlərindən biri molekulun qaynama və ya ərimə nöqtəsi kimi xüsusiyyətlərini proqnozlaşdırmaq ehtiyacıdır. Tədqiqatçılar bu proqnozu dəqiqləşdirdikdən sonra, dərmanlara, materiallara və daha çox şeyə gətirib çıxaran kəşflər verərək, işlərini davam etdirə bilirlər. Bununla belə, tarixən bu proqnozların açıqlanmasının ənənəvi üsulları əhəmiyyətli xərclərlə əlaqələndirilir – vəsaitdən əlavə vaxt və avadanlıqların köhnəlməsi.
Maşın öyrənməsi (ML) kimi tanınan süni intellekt sahəsinə daxil olun. ML molekul xüsusiyyətlərinin proqnozlaşdırılması yükünü bir qədər azaldıb, lakin yeni molekullar üçün sürətli proqnozlar vermək üçün mövcud məlumatlardan öyrənməklə prosesi ən effektiv surətdə sürətləndirən qabaqcıl vasitələr istifadəçidən əhəmiyyətli səviyyədə proqramlaşdırma təcrübəsi tələb edir. Bu, proqnozlaşdırma boru kəmərini idarə etmək üçün tələb olunan əhəmiyyətli hesablama biliyinə malik olmayan bir çox kimyaçılar üçün əlçatanlıq maneəsi yaradır.
Bu çətinliyi aradan qaldırmaq üçün MIT-dəki McGuire Tədqiqat Qrupunun tədqiqatçıları kimyaçılara qabaqcıl proqramlaşdırma bacarıqları tələb etmədən bu kritik proqnozları verməyə kömək edən istifadəçi dostu masa üstü proqramı olan ChemXploreML yaratdılar. Sərbəst mövcud olan, yükləmək asan və əsas platformalarda funksional olan bu proqram həm də tamamilə oflayn rejimdə işləmək üçün yaradılmışdır ki, bu da tədqiqat məlumatlarının mülkiyyətçi olmasına kömək edir.
Texnologiya Journal of Chemical Information and Modeling jurnalında bu yaxınlarda dərc edilmiş məqalədə təsvir edilmişdir .
Kimyəvi maşın öyrənməsində xüsusi maneələrdən biri molekulyar strukturları kompüterlərin başa düşə biləcəyi ədədi dilə çevirməkdir. ChemXploreML kimyəvi strukturları informativ ədədi vektorlara çevirən güclü, daxili “molekulyar yerləşdiricilər” ilə bu mürəkkəb prosesi avtomatlaşdırır. Daha sonra proqram intuitiv, interaktiv qrafik interfeys vasitəsilə nümunələri müəyyən etmək və qaynama və ərimə nöqtələri kimi molekulyar xassələri dəqiq proqnozlaşdırmaq üçün ən müasir alqoritmləri tətbiq edir.
McGuire Qrupunda postdok və məqalənin aparıcı müəllifi Aravindh Nivas Marimuthu deyir: “ChemXploreML-in məqsədi kimya elmlərində maşın öyrənməsindən istifadəni demokratikləşdirməkdir”.
“İntuitiv, güclü və oflayn işləyə bilən masa üstü tətbiqi yaratmaqla biz proqramlaşdırma mənşəyindən asılı olmayaraq ən müasir proqnozlaşdırıcı modelləşdirməni birbaşa kimyaçıların əlinə veririk. Bu iş yoxlama prosesini daha sürətli və ucuz etməklə nəinki yeni dərman və materialların axtarışını sürətləndirir, həm də onun çevik dizaynı yeniliklər üçün qapılar açır.”
ChemXploreML zaman keçdikcə təkamül etmək üçün nəzərdə tutulmuşdur, belə ki, gələcək texnika və alqoritmlər işlənib hazırlandıqca, tədqiqatçıların hər zaman ən müasir metodlara daxil olub həyata keçirə bilmələrini təmin edərək, onlar proqrama problemsiz inteqrasiya oluna bilər. Tətbiq üzvi birləşmələrin beş əsas molekulyar xassələri – ərimə nöqtəsi , qaynama nöqtəsi, buxar təzyiqi , kritik temperatur və kritik təzyiq üzrə sınaqdan keçirilmiş və kritik temperatur üçün 93%-ə qədər yüksək dəqiqlik əldə etmişdir.
Tədqiqatçılar həmçinin molekulları təmsil etmək üçün yeni, daha yığcam metodun (VICGAE) Mol2Vec kimi standart metodlar qədər dəqiq olduğunu, lakin 10 dəfəyə qədər daha sürətli olduğunu nümayiş etdirdilər.
Marimuthu deyir: “Biz istənilən tədqiqatçının dayanıqlı materialların hazırlanmasından tutmuş ulduzlararası kosmosun mürəkkəb kimyasını tədqiq etməyə kimi unikal problemləri həll etmək üçün maşın öyrənməsini asanlıqla fərdiləşdirə və tətbiq edə biləcəyi bir gələcəyi nəzərdə tuturuq”. Kağızda ona qoşulan baş müəllif və 1943-cü il Karyera İnkişafı üzrə Dosent Kimya professoru Brett McGuire idi.
Daha çox məlumat: Aravindh Nivas Marimuthu et al, ChemXploreML-dən istifadə edərək Molekulyar Mülkiyyətin Proqnozlaşdırılması üçün Maşın Öyrənmə Boru Kəməri, Kimyəvi Məlumat və Modelləşdirmə jurnalı (2025). DOI: 10.1021/acs.jcim.5c00516
Jurnal məlumatı: Kimyəvi İnformasiya və Modelləşdirmə jurnalı
Massaçusets Texnologiya İnstitutu tərəfindən təmin edilmişdir

