



Maşın öyrənməsi kvant kimyasının mərkəzi problemini həll etməyə kömək edir
Marietta Fuhrmann-Koch, Heidelberg Universiteti
Stephanie Baum tərəfindən redaktə edilib , Robert Egan tərəfindən nəzərdən keçirilib
Tercih edilən mənbə kimi əlavə edin
Kredit: Amerika Kimya Cəmiyyətinin Jurnalı (2025). DOI: 10.1021/jacs.5c06219
Heydelberq Universitetinin alimləri kvant kimyası tədqiqatlarına yeni maşın öyrənmə metodlarını tətbiq etməklə hesablama kimyasında əhəmiyyətli irəliləyişlər əldə ediblər. Onlar kvant kimyasında onilliklər boyu davam edən bir dilemmanın həllində böyük bir irəliləyiş əldə ediblər: molekulyar enerjilərin və elektron sıxlığının dəqiq və sabit hesablanması, xeyli az hesablama gücündən istifadə edən və buna görə də çox böyük molekullar üçün hesablamalara imkan verən sözdə orbitalsız yanaşma ilə.
STRUCTURES Mükəmməllik Klasterində, Elmi Hesablama üzrə Fənlərarası Mərkəzin (IWR) iki tədqiqat qrupu, uzun müddət etibarsız hesab edilən hesablama prosesini dəqiq nəticələr verməsi və fiziki cəhətdən mənalı bir həll yolu təmin etməsi üçün təkmilləşdiriblər. Nəticələr Amerika Kimya Cəmiyyətinin Jurnalında dərc olunub .
Molekulyar elektron sıxlığı niyə vacibdir
Bir molekulda elektronların necə paylanması onun kimyəvi xüsusiyyətlərini – sabitliyindən və reaktivliyindən tutmuş bioloji təsirinə qədər müəyyən edir. Bu elektron paylanmasını və nəticədə yaranan enerjini etibarlı şəkildə hesablamaq kvant kimyasının əsas funksiyalarından biridir. Bu hesablamalar, molekulların xüsusi olaraq başa düşülməli və dizayn edilməli olduğu bir çox tətbiqin əsasını təşkil edir, məsələn, yeni dərmanlar, daha yaxşı batareyalar, enerji çevrilməsi üçün materiallar və ya daha səmərəli katalizatorlar üçün.
Lakin bu cür hesablamalar hesablama baxımından çox mürəkkəbdir və tez bir zamanda çox mürəkkəbləşir. Molekul nə qədər böyük olarsa və ya yoxlanılması lazım olan variantlar nə qədər çox olarsa, bir o qədər tez müəyyən edilmiş hesablama prosesləri öz həddinə çatar. “Orbitallarsız Kvant Kimyası” layihəsi burada kimya, fizika və süni intellekt tədqiqatlarının kəsişməsində yerləşir.
Orbital sıxlıq funksional nəzəriyyəsinin canlanması
Kvant kimyasında molekullar tez-tez sıxlıq funksional nəzəriyyəsi ilə təsvir olunur ki, bu da kvant mexaniki dalğa funksiyasını hesablamadan kimyəvi molekulyar xüsusiyyətlərin fundamental proqnozlaşdırılmasına imkan verir. Bunun əvəzinə elektron sıxlığı əsas kəmiyyət kimi istifadə olunur ki, bu da hesablamaları nəhayət praktik hala gətirən sadələşdirmədir. Bu orbitalsız yanaşma xüsusilə səmərəli hesablamalar vəd edir, lakin indiyə qədər elektron sıxlığındakı kiçik sapmalar qeyri-sabit və ya “qeyri-fiziki” nəticələrə səbəb olduğundan, o qədər də faydalı hesab edilmirdi.
Maşın öyrənməsinin köməyi ilə Heidelberg metodu nəhayət ki, bir çox müxtəlif üzvi molekullar üçün bu dəqiqlik və sabitlik problemini həll edir.
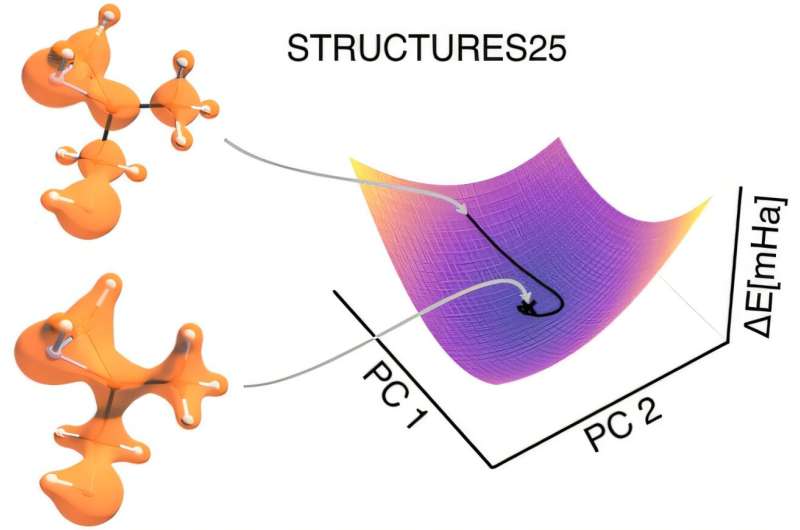
STRUCTURES25 modeli necə işləyir
STRUCTURES25 adlanan yeni proses, elektron sıxlığı və enerji arasındakı əlaqəni dəqiq istinad hesablamalarından birbaşa öyrənən və hər bir atomun kimyəvi mühitini riyazi cəhətdən ətraflı təsvirdə əks etdirən xüsusi olaraq hazırlanmış neyron şəbəkəsinə əsaslanır. Unikal təlim konsepsiyası əsas idi: Model təkcə konvergent elektron sıxlıqları ilə deyil, həm də əsas istinad hesablamalarında hədəflənmiş, idarə olunan dəyişikliklər nəticəsində yaranan düzgün həlli əhatə edən bir çox variantla öyrədildi.
Buna görə də, bu hesablama prosesi, hətta kiçik sapmalar halında belə, molekulyar enerjilər və elektron sıxlığı üçün fiziki cəhətdən mənalı bir həll yolu tapa bilir. Heidelberg tədqiqatçıları vurğulayırlar ki, hesablamada “yolunu itirmədən” sabit qalır.
Mürəkkəb, dərmana bənzər molekullar üzərində performans
Böyük və müxtəlif üzvi molekullar kolleksiyası üzərində aparılan sınaqlarda STRUCTURES25, ilk dəfə olaraq orbitalsız yanaşmadan istifadə edərək sabit bir konvergensiya nümayiş etdirərək, müəyyən edilmiş istinad hesablamaları ilə rəqabət apara biləcək bir dəqiqliyə nail oldu. Metodun performansı yalnız kiçik nümunələrdə deyil, həm də xeyli böyük ” dərman kimi” molekullarda da nümayiş etdirildi.
İlkin işləmə müddəti müqayisələri sübut edir ki, hesablama prosesi molekul ölçüsünün artması ilə daha yaxşı miqyaslana bilər və beləliklə, hesablama sürətini artıra bilər. Əvvəllər çox mürəkkəb hesab edilən hesablamalar artıq əlçatandır.
Gündəlik məlumat üçün Phys.org-a etibar edən 100.000-dən çox abunəçi ilə elm, texnologiya və kosmosdakı ən son yenilikləri kəşf edin . Pulsuz bülletenimizə abunə olun və vacib olan nailiyyətlər, innovasiyalar və tədqiqatlar haqqında gündəlik və ya həftəlik yeniliklərdən xəbərdar olun .
Daha sürətli, miqyaslana bilən simulyasiyalar üçün nəticələr
IWR-də Elmi Süni İntellekt tədqiqat qrupuna rəhbərlik edən professor Dr. Fred Hamprext bildirir ki, “Orbital sıxlıqsız funksional nəzəriyyə uzun müddətdir daha sürətli hesablama vədini daşıyırdı – lakin xahiş edirəm fizika hesabına deyil.” “STRUCTURES25 ilə biz ilk dəfə olaraq hesablamanın həm kimyəvi cəhətdən dəqiq enerjiləri, həm də elektron sıxlığının sabit, praktik optimallaşdırılmasını əhatə edə biləcəyini nümayiş etdiririk.”
IWR-də Nəzəri və Hesablama Kimyası tədqiqat qrupunun rəhbəri professor Dr. Andreas Dreuw əlavə edir: “Optimallaşdırma artıq qeyri-sabit deyil və buna görə də yüksək dəqiqliklə xeyli sürətli proqnozlar üçün irəliyə doğru böyük bir addımdır. İndi simulyasiyalar klassik proseslərin çətinliklə toxuna biləcəyi məsafədədir, məsələn, bir çox konfiqurasiyanın və ya çox böyük molekulların araşdırılmasına ehtiyac olduqda.”
Nəşr detalları
Roman Remme və digərləri, Maşın Öyrənməsi ilə Gücləndirilmiş Sabit və Dəqiq Orbital Sıxlıq Funksional Nəzəriyyəsi, Amerika Kimya Cəmiyyətinin Jurnalı (2025). DOI: 10.1021/jacs.5c06219
Jurnal məlumatı: Amerika Kimya Cəmiyyətinin Jurnalı
Heydelberq Universiteti tərəfindən təmin edilir